Run samtools view on WES with HTTP URLs as input
In the previous step, we configured and started instances of WES, DRS, and the Starter Kit UI.
Workflow and Dataset
We will now attempt to run a Nextflow-based samtools view workflow to count the number of reads in BAM files. We will use some small BAMs from the Tabula Muris open dataset as our test dataset.
Note: For reference, you may wish to read more about Tabula Muris, view the Github Repo, and see more info about the files uploaded to the Tabula Muris s3 bucket.
The Nextflow workflow is version controlled on Github at https://github.com/jb-adams/samtools-view-count-nf. We can tell WES to run this workflow simply by providing this Github repository URL.
Submit workflow run request to WES
The samtools-view-count-nf takes one input parameter, --input, which must reference a local (file path) or remote (URL) SAM/BAM/CRAM file. As our Tabula Muris test dataset BAMs are all stored on a public AWS S3 bucket, we can submit these as URLs to the workflow.
For now, let's work with the following BAM URL:
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam
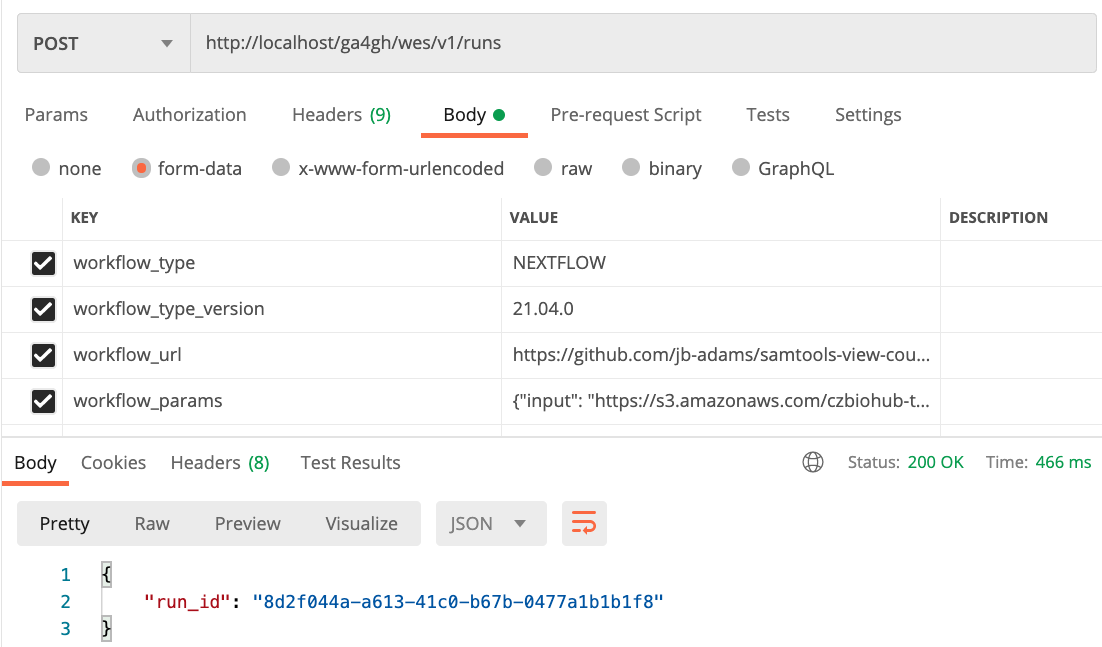
Let's submit a workflow run request to WES, running our samtools view workflow on the above input BAM. To do so, issue the following HTTP request using your preferred API testing tool:
Method and URL
POST http://localhost/ga4gh/wes/v1/runs
Headers
Content-Type: multipart/form-data
Form data (body)
workflow_type: NEXTFLOW
workflow_type_version: 21.04.0
workflow_url: https://github.com/jb-adams/samtools-view-count-nf
workflow_params: {"input": "https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam"}
Upon doing so, we should receive a single run_id, indicating that the workflow run request was accepted. The run_id provided to us will allow us to monitor the status of the workflow run. In this example, a run_id of 8d2f044a-a613-41c0-b67b-0477a1b1b1f8 was returned, however each time a run request is submitted to WES, and randomly generated run_id will be returned.
Example workflow run request and run_id response:
Monitor workflow run
If we give the workflow run a minute to complete, we can monitor its status by passing the run_id to the GET /runs/{run_id} endpoint:
Example request (substitute in your own run_id):
GET http://localhost/ga4gh/wes/v1/runs/8d2f044a-a613-41c0-b67b-0477a1b1b1f8
Response:
{
"run_id": "8d2f044a-a613-41c0-b67b-0477a1b1b1f8",
"request": {
"workflow_params": {
"input": "https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam"
},
"workflow_type": "NEXTFLOW",
"workflow_type_version": "21.04.0",
"workflow_url": "https://github.com/jb-adams/samtools-view-count-nf"
},
"state": "COMPLETE",
"run_log": {
"name": "jb-adams/samtools-view-count-nf",
"cmd": [
"#!/bin/bash -ue",
"echo \"Running samtools view on https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam\" >&2",
"samtools view -c https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam"
],
"start_time": "2021-09-03T13:40:06Z",
"end_time": "2021-09-03T13:40:20Z",
"stdout": "http://localhost/ga4gh/wes/v1/logs/nextflow/stdout/8d2f044a-a613-41c0-b67b-0477a1b1b1f8?workdirs=0f%2F4340a1a21885f9bfb182bf34a85d2f",
"stderr": "http://localhost/ga4gh/wes/v1/logs/nextflow/stderr/8d2f044a-a613-41c0-b67b-0477a1b1b1f8?workdirs=0f%2F4340a1a21885f9bfb182bf34a85d2f",
"exit_code": 0
},
"task_logs": [
{
"name": "samtools_view",
"cmd": [
"#!/bin/bash -ue",
"echo \"Running samtools view on https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam\" >&2",
"samtools view -c https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam"
],
"start_time": "2021-09-03T13:40:06Z",
"end_time": "2021-09-03T13:40:20Z",
"stdout": "http://localhost/ga4gh/wes/v1/logs/nextflow/stdout/8d2f044a-a613-41c0-b67b-0477a1b1b1f8/0f/4340a1a21885f9bfb182bf34a85d2f",
"stderr": "http://localhost/ga4gh/wes/v1/logs/nextflow/stderr/8d2f044a-a613-41c0-b67b-0477a1b1b1f8/0f/4340a1a21885f9bfb182bf34a85d2f",
"exit_code": 0
}
],
"outputs": {}
}
The above response payload tells us information about our workflow run:
state: COMPLETEindicates the workflow run completed successfully- the
requestattribute indicates the initial request parameters we supplied to trigger the workflow run run_loggives us a summary of the entire run, including start and end times, stdout and stderr outputoutputsprovides URLs/paths to any output files/directories created during the workflow. In this case, no output files were created, all output is available instdout
If we follow the URL at run_log.stdout, we will find the main result of the workflow run, that is, the read count of the input BAM file.
For example:
GET http://localhost/ga4gh/wes/v1/logs/nextflow/stdout/8d2f044a-a613-41c0-b67b-0477a1b1b1f8?workdirs=0f%2F4340a1a21885f9bfb182bf34a85d2f
Returns a response of:
848036
Therefore, the BAM file at https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam contains 848036 reads.
Run workflow with different inputs
Try running the same workflow with different input BAMs. Use WES API calls to submit the workflow run request and monitor its output (the read count). Below are some input URLs and expected read counts you can use to validate the run was configured correctly and completed successfully.
| Input URL | Read Count |
|---|---|
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R1.mus.Aligned.out.sorted.bam | 848036 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000126-3_39_F-1-1_R2.mus.Aligned.out.sorted.bam | 806872 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000127-3_38_F-1-1_R1.mus.Aligned.out.sorted.bam | 821666 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/A1-B000127-3_38_F-1-1_R2.mus.Aligned.out.sorted.bam | 821604 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/B7-MAA000586-3_8_M-1-1_R1.mus.Aligned.out.sorted.bam | 3950888 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/C8-B002433-3_38_F-1-1_R1.mus.Aligned.out.sorted.bam | 1539546 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/D5-MAA000452-3_8_M-1-1_R1.mus.Aligned.out.sorted.bam | 3912618 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/E20-MAA000652-3_10_M-1-1_R1.mus.Aligned.out.sorted.bam | 758692 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/F17-B002765-3_38_F-1-1_R1.mus.Aligned.out.sorted.bam | 998700 |
https://s3.amazonaws.com/czbiohub-tabula-muris/facs_bam_files/F19-D041912-3_8_M-1-1_R1.mus.Aligned.out.sorted.bam | 1521586 |
Summary
In this section, we ran the samtools view workflow to count reads in BAM files by executing simple HTTP requests to the WES service. You may have noticed that we did not need DRS or the UI for this section. In the next section, we will explore how to run the same workflow with DRS URLs instead of direct HTTP URLs as input, and how this abstraction provides us with much more flexibility in terms of where input resources can be stored.